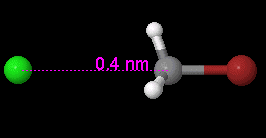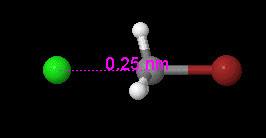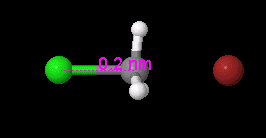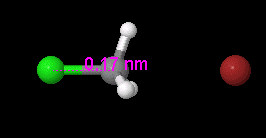
View, compare, convert 3D chemical structures, even PyMOL, with Jmol viewer
Jmol is an open-source viewer for three-dimensional chemical structures, with features for chemicals, crystals, materials and biomolecules. It can read many file types, including PDB, CIF, SDF, MOL, PyMOL PSE files, and Spartan files, as well as output from Gaussian, GAMESS, MOPAC, VASP, CRYSTAL, CASTEP, QuantumEspresso, VMD, and many other quantum chemistry programs.
Files can be transferred directly from several databases, including RCSB, EDS, NCI, PubChem, and MaterialsProject.
Multiple files can be loaded and compared. A scripting language and a web API allow customization of the user interface. Jmol interfaces with JSpecView for spectroscopy, JSME for 2D->3D conversion, POV-Ray for images, and CAD programs for 3D printing (VRML export).
Features:
World-wide user group * Low footprint option (50K) for simple interactive structure display * Full crystallographic symmetry capability * Many surface formats, surfaces on the fly * Reads over 60 file formats, including PyMOL (PSE) session files * Creates highly compressed (300:1) surface files from volumetric (CUBE) data * Exports to GIF, JPG, PNG, PDF, WRL, POV-Ray, OBJ formats * Reading of JCAMP-DX, CML, AnIML formats * Interactive real and predicted 1H NMR spectra * Interactive IR, Raman, NMR, GC/MS, UV/VIS spectra * Spectra generated in PDF format, and more.